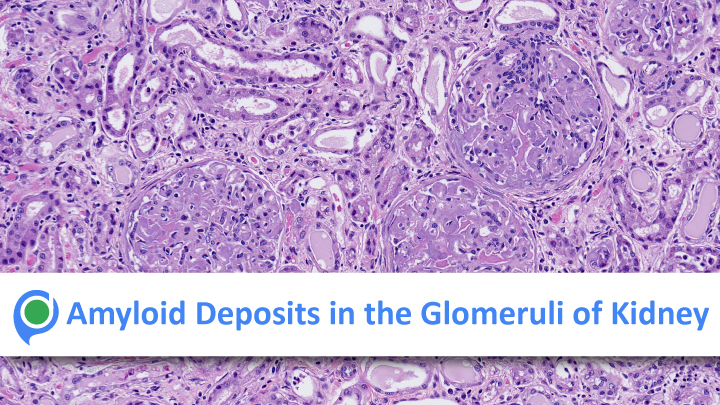
Renal amyloidosis characterized by the pathologic deposition within glomeruli and/or interstitium of congophilic fibrils mostly comprise of either immunoglobulin light chains or serum amyloid A related protein and, less commonly, mutated forms of apolipoproteins AI or AII, lysozyme, fibrinogen, gelsolin, or transthyretin. Amyloidosis is characterized by extracellular deposition of insoluble fibrils, which results from the abnormal folding of proteins. Amyloidosis can either be localized or systemic and may affect any organ.
The kidney is the most commonly affected organ in systemic amyloidosis. The two most common types of renal amyloidosis are immunoglobulin light chain-derived amyloidosis and reactive (secondary) amyloidosis, but several of the rare hereditary forms of amyloidosis, such as those derived from fibrinogen A, apolipoprotein AI, apolipoprotein AIV, transthyretin, gelsolin,…